软件教程
来自我们的高级技术支持专家,精心撰写整理的原创学习教程
原创精选
便捷搜索
快速入门和提高
教程(六)GMMX 构象搜索模块使用方法
发布时间:2019-04-08
关键词:GMMX 构象搜索模块
(一)引言:
➢ GaussView 6 可以使用新增GMMX模块进行构象搜索。
➢ 一旦GMMX 搜索完成,所有的构象信息将会列出,你可以使用GaussView 6以单个分子组形式打开和查看。同样,你可以查看这些构象的能量。对于大部分研究者来说这是一个非常重要的功能。
➢ 本文将以丙三醇(甘油)为例,讲解如何使用GMMX进行构象搜索,继而使用第一性原理方法进行优势构象筛选。
(二)计算过程:
1. 分子建模
█ 软件版本Gaussian 16, GaussView 6。使用GaussView 6 构建丙三醇分子,如下图:
█ 下图左边是构建的初始结构,为了有个良好的初始构型,先使用半经验量子力学PM6方法对其进行结构优化,得到较好的初始构型(下图右)。可以看出,经过PM6方法优化后的结构和最开始画的结构(下图左)有明显区别。
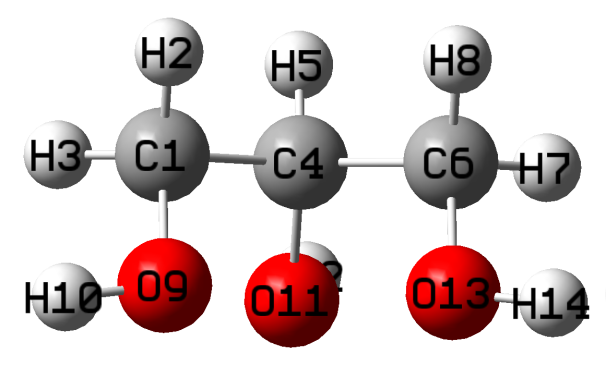
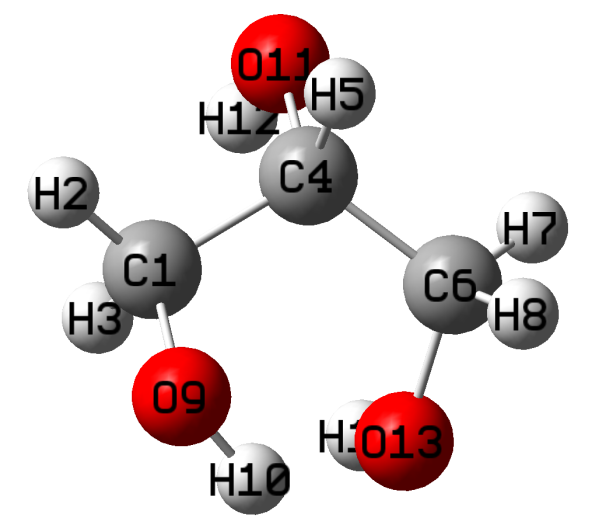
丙三醇的初始结构 PM6优化后的丙三醇结构
█ 打开GMMX面板
◆ 看到如下画面:
◆ “力场”选项里有三种力场,默认力场是MMFF94,这是一个通用的有机力场,主要设计用于小的类药物有机分子。MMFF力场基于MM3力场,但并没有针对一种用途进行优化,例如模拟蛋白质或小分子,其在有机化学计算中表现均较好。另外两个力场(MMX和MM3)来自Allinger组,与MMFF94在延长共轭处理上有所不同。
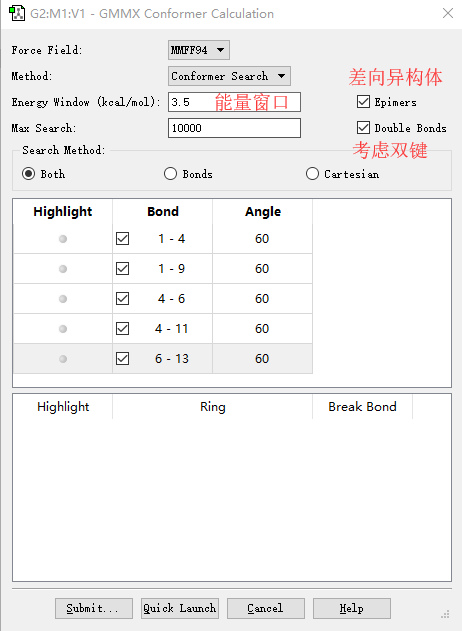
GMMX界面及功能
█ Method里默认的功能就是构象搜索。能量窗口默认的是3.5 kcal/mol,表示寻找的最低能量构象和最高能量构象的差值小于3.5 kcal/mol。如果勾选Epimers和 Double Bonds就代表考虑差向异构体和双键的转动。
◆ 下半部分就是索要旋转的键,可以用Highlight高亮显示出来。
2. 构象搜索计算
█ 上图中的参数选好之后,就可以点击submit进行构象搜索了。
◆ 构象搜索的计算时间随着体系的增大而变长,有些大分子需要计算较长时间。
◆ 计算好之后GaussView 6 将显示以下界面:
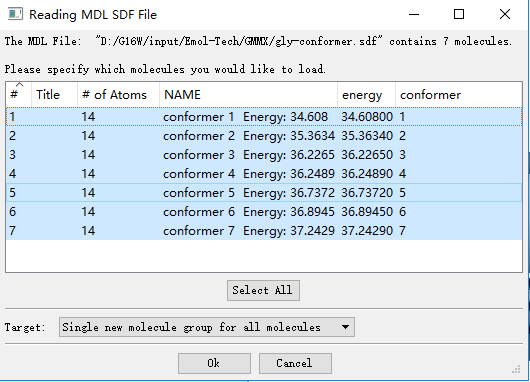
█ 打开gly-conformer.sdf,点击Select All得到一系列构象,如下图所示:
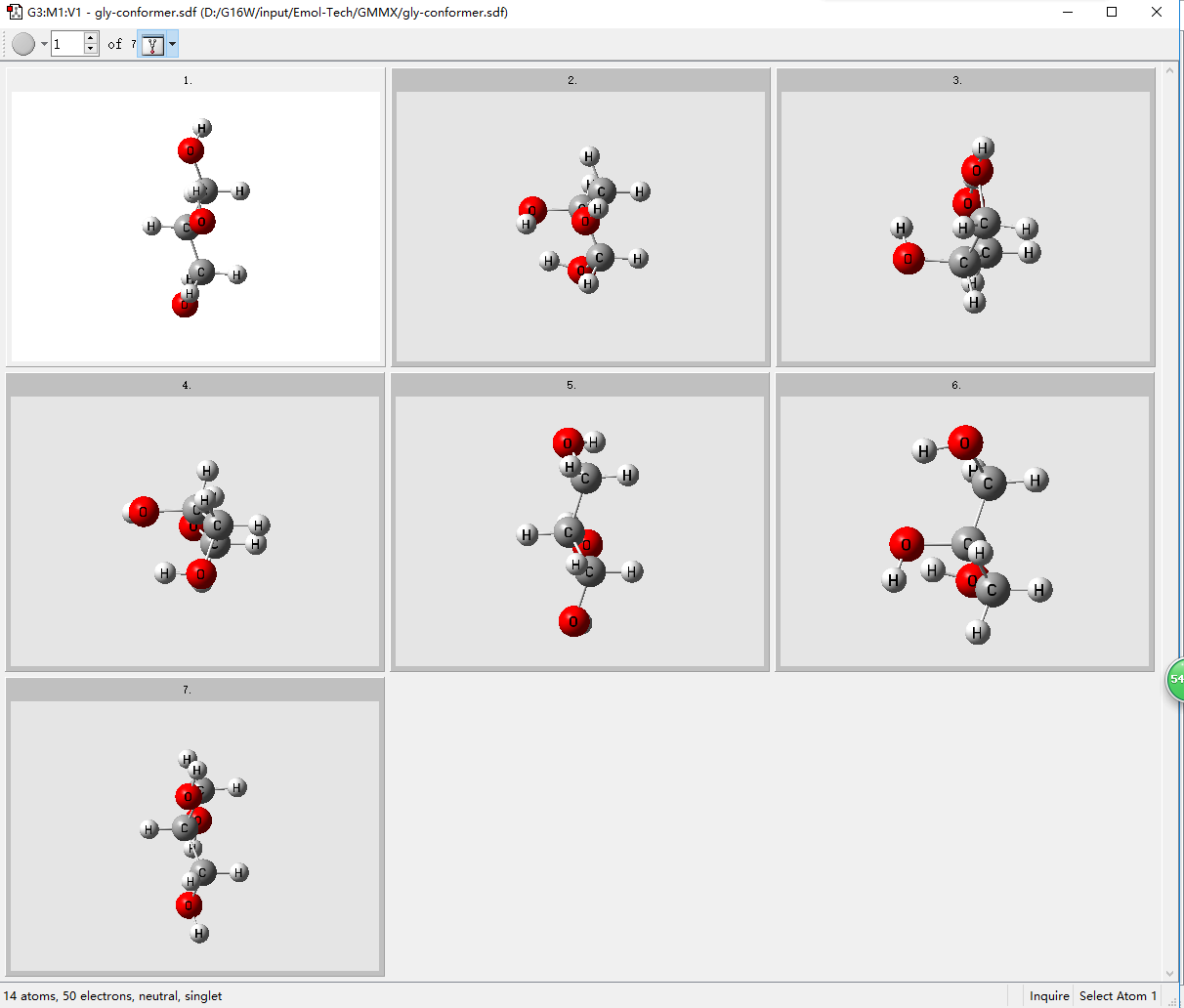
丙三醇构象搜索结果文件及所有构象
3. 能量曲线
█ 打开GaussView 6 中的Results → Energy Plot,得到上述7个构象的能量曲线。
█ 可以看到这7个构象能量相差在3.5 kcal/mol以内。
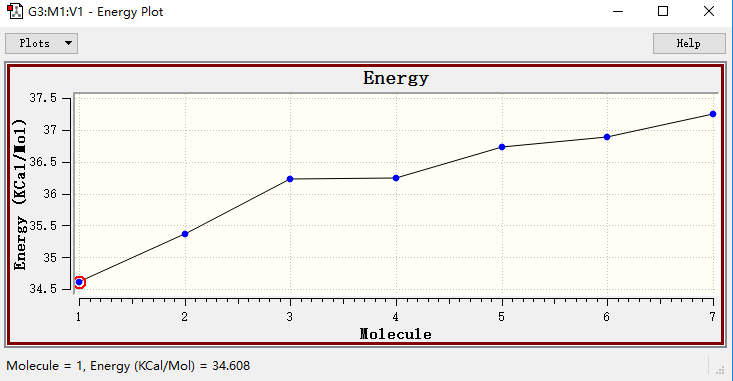
丙三醇构象能量曲线图
4. 使用Gaussian 16 中的DFT方法进行最终构象确定(B3LYP/6-31G(d))
█ 批量编辑输入文件
◆ 设置好计算方法后,点击Assign to Molecule group
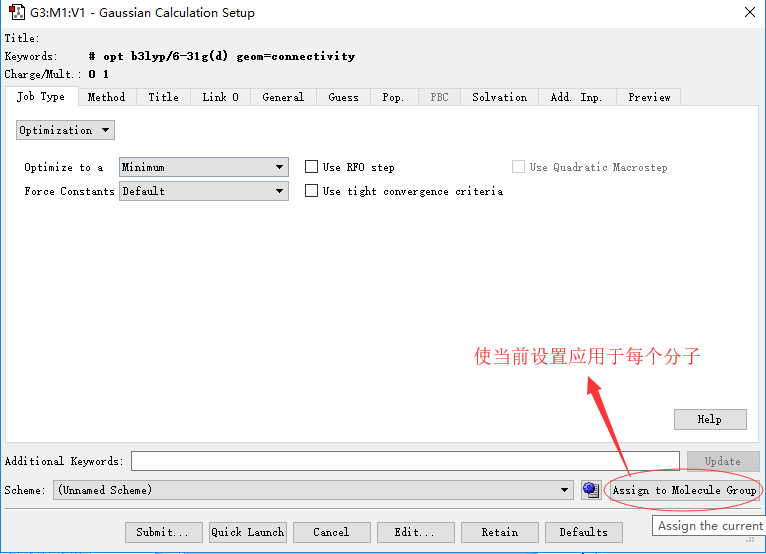
输入文件的批量编辑(a)
◆ 点击submit选项提交,会让你保存输入文件,此时点击 Advanced,出现如下界面:
◆ 在Save Molecule Group选项中选择“Yes, separate file for each molecule”,给每个构象建立独立文件;此外在Actions选项中选择给每个构象文件增加一个独立编号,这样所有的构象就有一一对应的输入文件了。
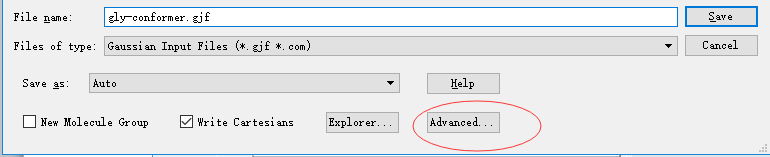
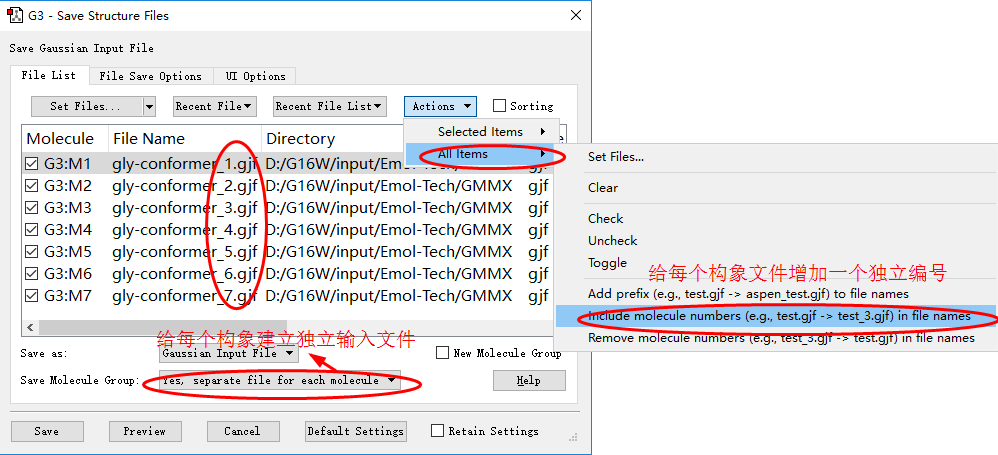
输入文件的批量编辑(b)
█ Gaussian 16计算
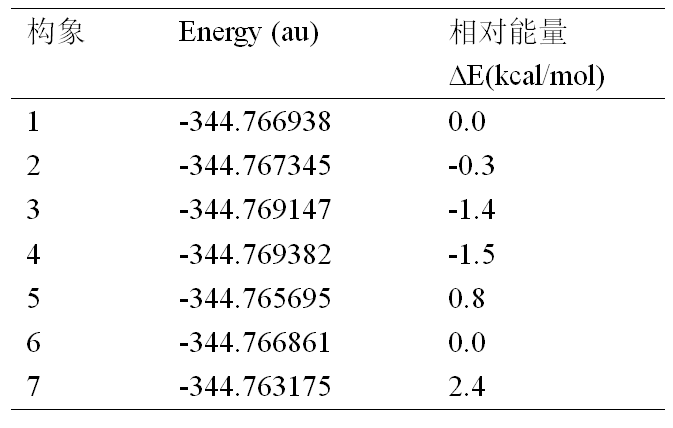
◆ 把所建立的7个输入文件提交给Gaussian 16进行优化,然后用GaussView 6打开,获得7个构象的最终能量如下。可以看出,最稳定结构是4号构象。
◆ 到此为止就完成了GMMX联合第一性原理计算获得化合物最稳定构象的工作
(三)知识背景:
➢ 分子力场介绍
█ https://en.wikipedia.org/wiki/Merck_Molecular_Force_Field
█ 更多参考可以点击GMMX界面下的HELP功能。
➢ 能量窗口的选择需要根据实际情况来选择,比如体系所处的温度、压力和化学环境等。
➢ 由于当前的提供的分子力场主要基于有机化学体系,因此在含有金属的体系使用时要格外小心,可以通过选择合适的键、键角、二面角来避开金属原子的干扰。