软件教程
来自我们的高级技术支持专家,精心撰写整理的原创学习教程
原创精选
便捷搜索
快速入门和提高
教程(八)红外光谱模拟
发布时间:2019-05-10
关键词:红外光谱模拟
(一)引言:
➢ 计算化学中一个很重要的内容就是各类谱图的模拟。红外光谱则是计算化学中最常见的可以模拟的光谱。本章内容以丙烯(C3H6)分子为例,介绍如何进行红外光谱的预测及谱峰如何指认。
(二)计算过程:
1. 模型建立
█ 软件版本Gaussian 16, GaussView 6
█ 使用方法:B3LYP/6-311G(d,p)。计算设置如下图所示:
◆ 红外光谱的计算设置很简单,只要选择Job Type中的Opt+Freq,然后在Method面板选择好方法和基组即可。
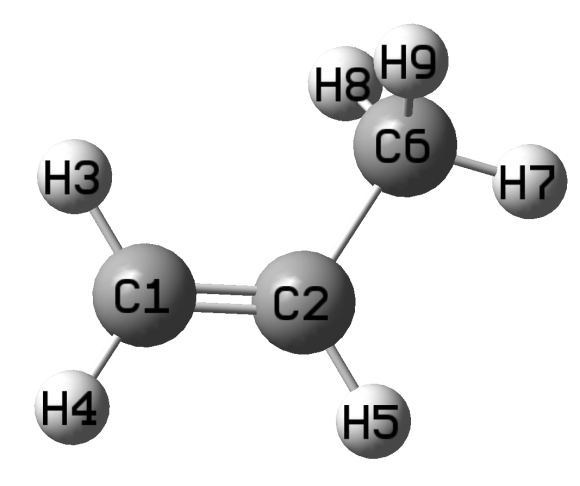
丙烯分子结构

红外光谱计算设置
2. 红外光谱计算
█ 设置好计算参数后,保存为c3h6.gjf,然后交给Gaussian 16进行计算。
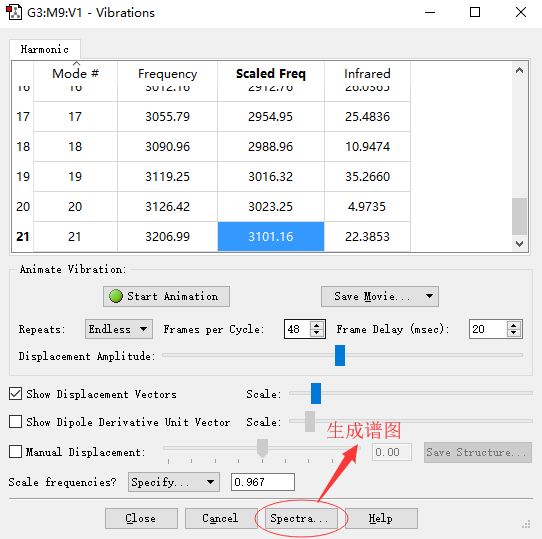
█ 打开计算好的c3h6.log文件,然后选择Resultsvibrations,则会打开振动光谱面板。
█ 下图中,我们需要使用校正因子让振动频率更加接近实验值。本文中使用的方法B3LYP/6-311G(d,p)对应的校正因子为0.967。更多信息请参考“知识背景”部分。
█ 设置好校正因子后,我们可以得到如图所示的校正后的频率。
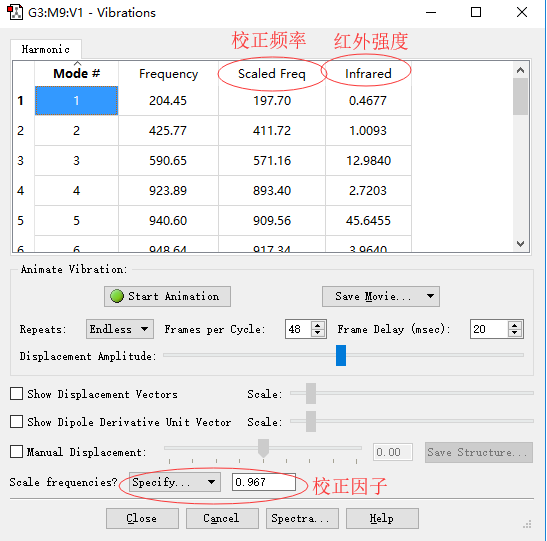
振动光谱面板
3. 振动模式分析
█ 完成上述设置后,点击Start Animation以及勾选Show Displacement Vectors。然后点击每个频率,都会出现相应的振动模式。比如此处我们点击第21个振动模式,对应的频率为3101.16 cm-1,其对应的振动模式如下图所示。
█ 同理,所有的振动频率对应的振动模式都可以得到。
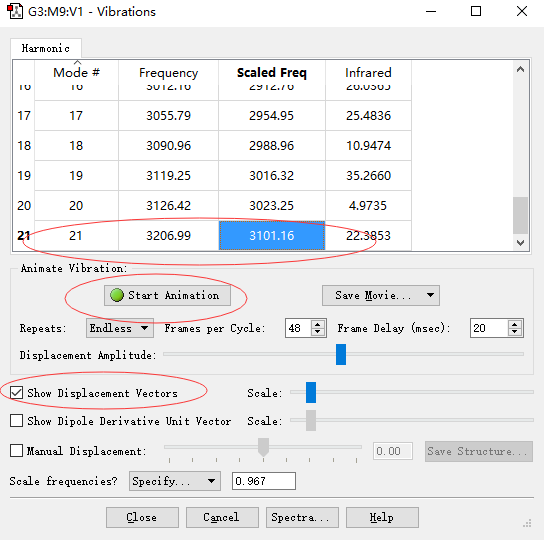
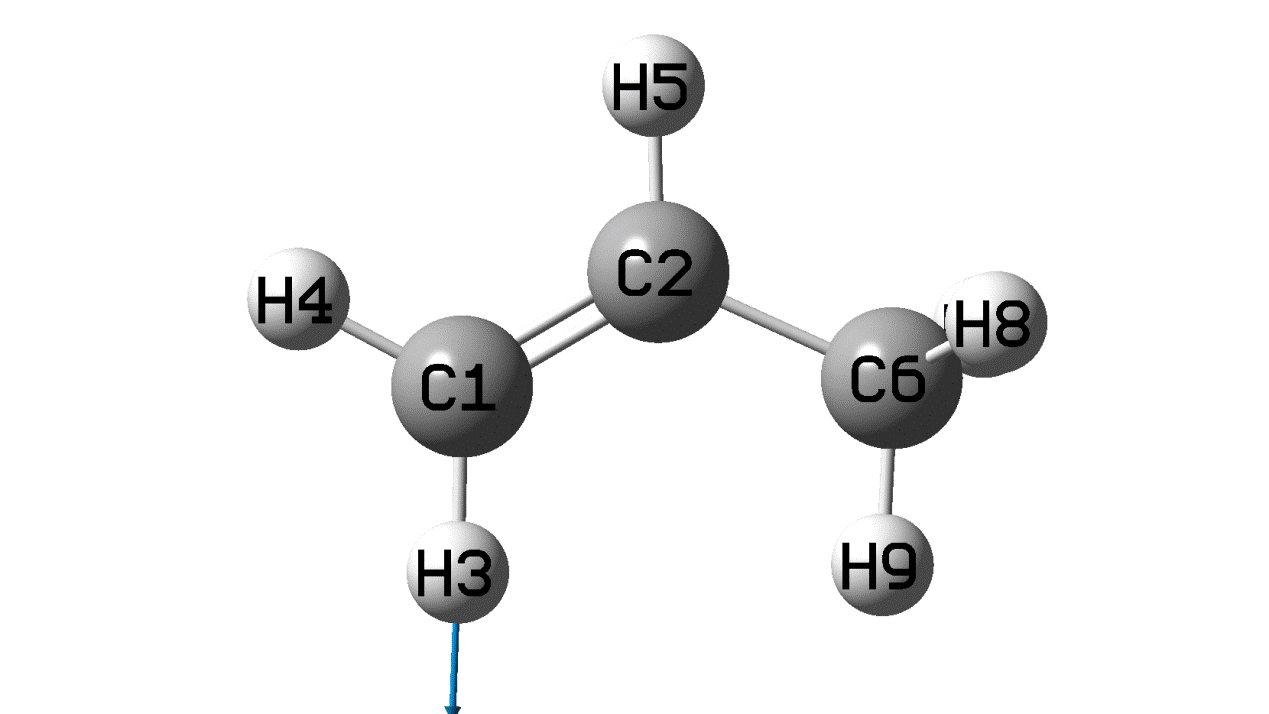
丙烯分子-CH2基团的非对称伸缩振动
4. 谱峰指认
█ 此时点击振动面板中的Spectra,则会生成预测的红外光谱图。如下图所示
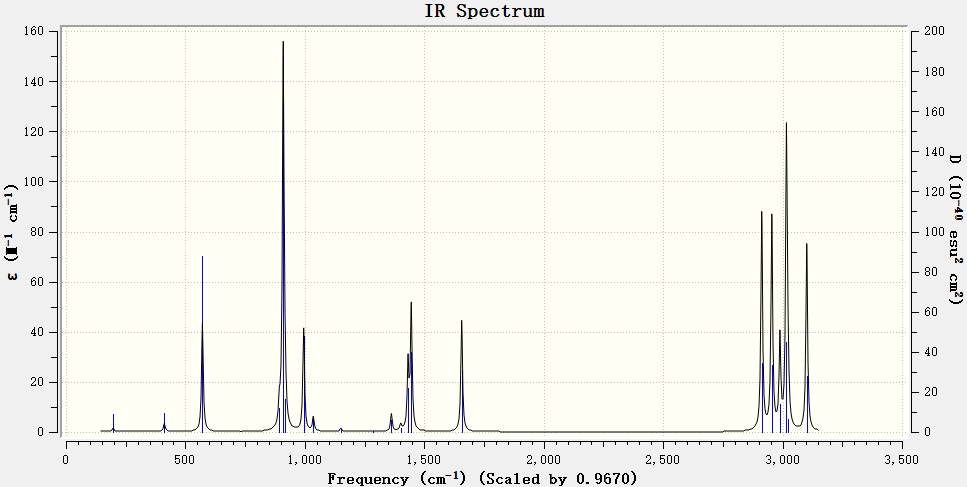
█ 仔细观察可以发现,默认红外光谱图坐标与常用实验红外光谱坐标相反,所以在IR谱图上点击右键中的Property将坐标反转,得到下图。

反转坐标
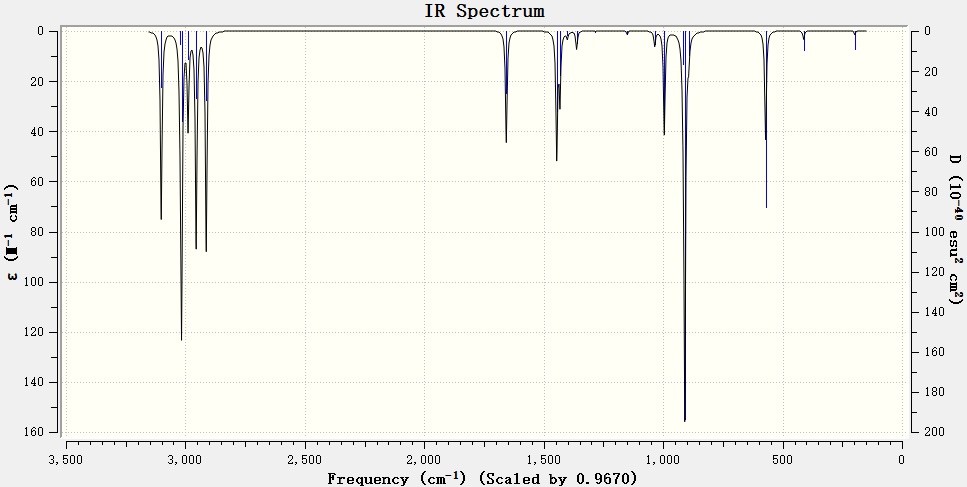
█ 丙烯分子气态红外光谱可以从NIST数据库获得。更多信息请参考“知识背景”部分。
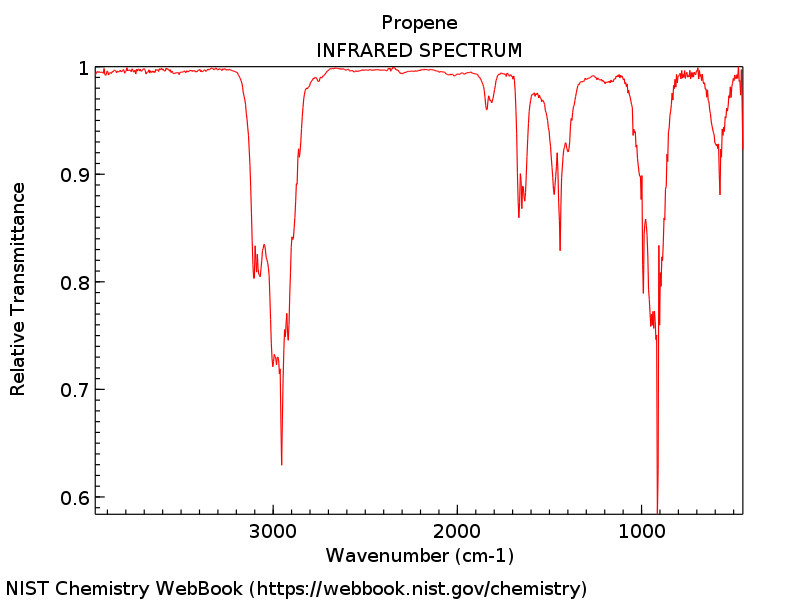
丙烯分子实验红外光谱图
█ 对比实验红外光谱和理论预测的红外光谱,可以看出理论预测结果于实验值吻合较好。
█ 然后可以通过点击谱峰位置,得到每个谱峰所对应的振动模式,即完成了谱峰指认。
(三)知识背景:
➢ 关于振动分析更多内容可以参考Gaussian主页:http://gaussian.com/vib/
➢ 推荐书籍:Quantum Chemistry, by Levine.
➢ 频率校正因子网页:https://cccbdb.nist.gov/vibscale.asp
➢ NIST数据库:https://webbook.nist.gov/chemistry/